PAPRECA flowchart
The main operations of PAPRECA can be summarized in the following flowchart:
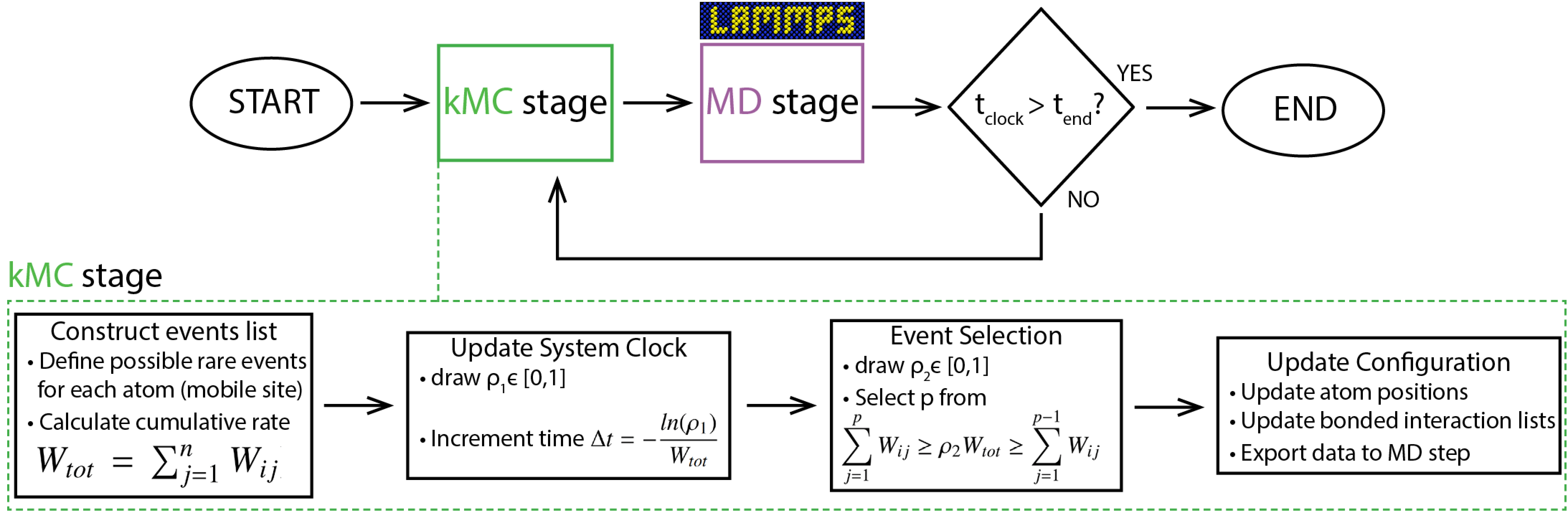
Note that, in the current version of PAPRECA, event scanning is parallelized. However, event execution is performed serially. More specifically, after discovering all local events, the total rate of each MPI process is communicated to the master MPI processes (i.e., proc 0). Firstly, the master process calculates the total rate of the current step and advances the simulation clock:
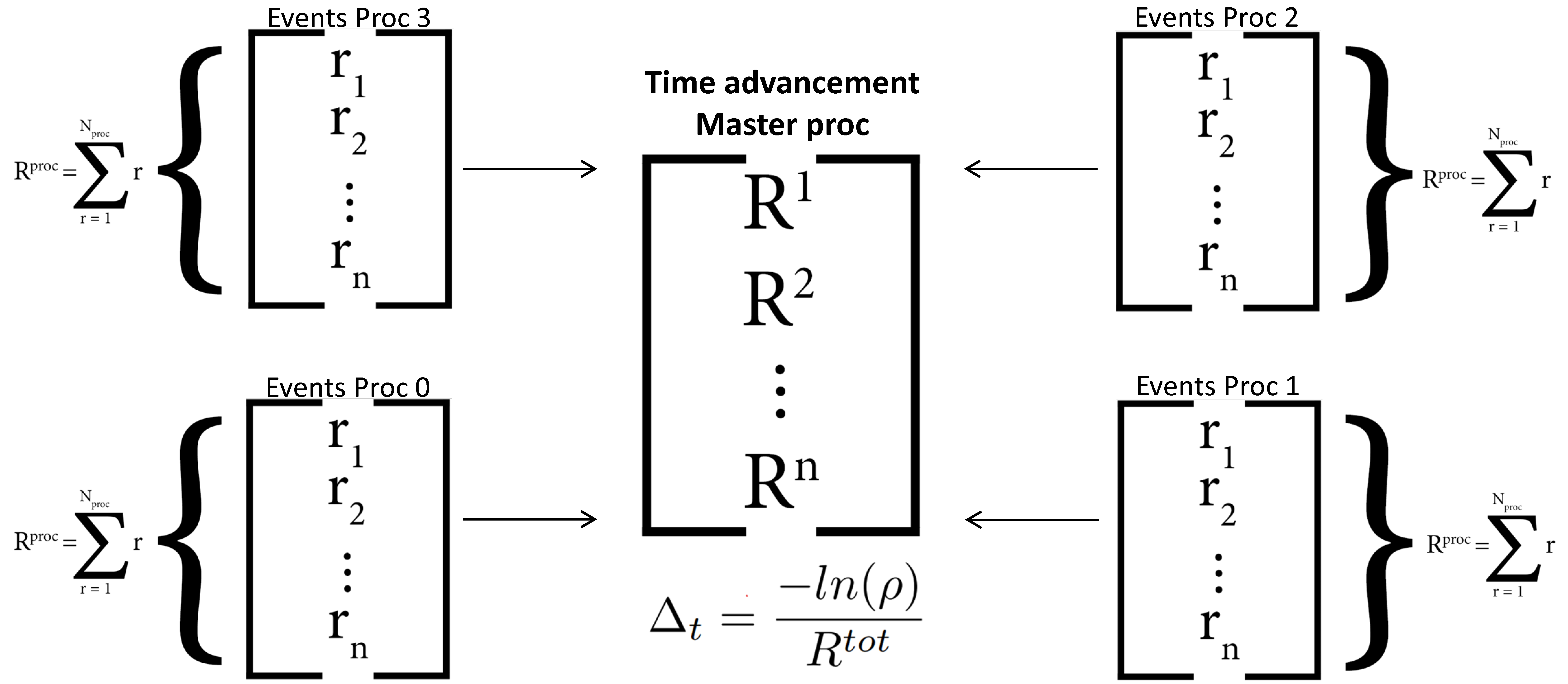
Where Δt is the kMC time interval, ρ is a pseudo-random uniformly distributed random number between 0 and 1, R is the per-MPI-process/total rate on a given step, and r is a local event rate. After advancing the simulation clock, the master MPI process chooses the event MPI processor by performing a (rejection-free) kMC selection [1], [2], [3]:
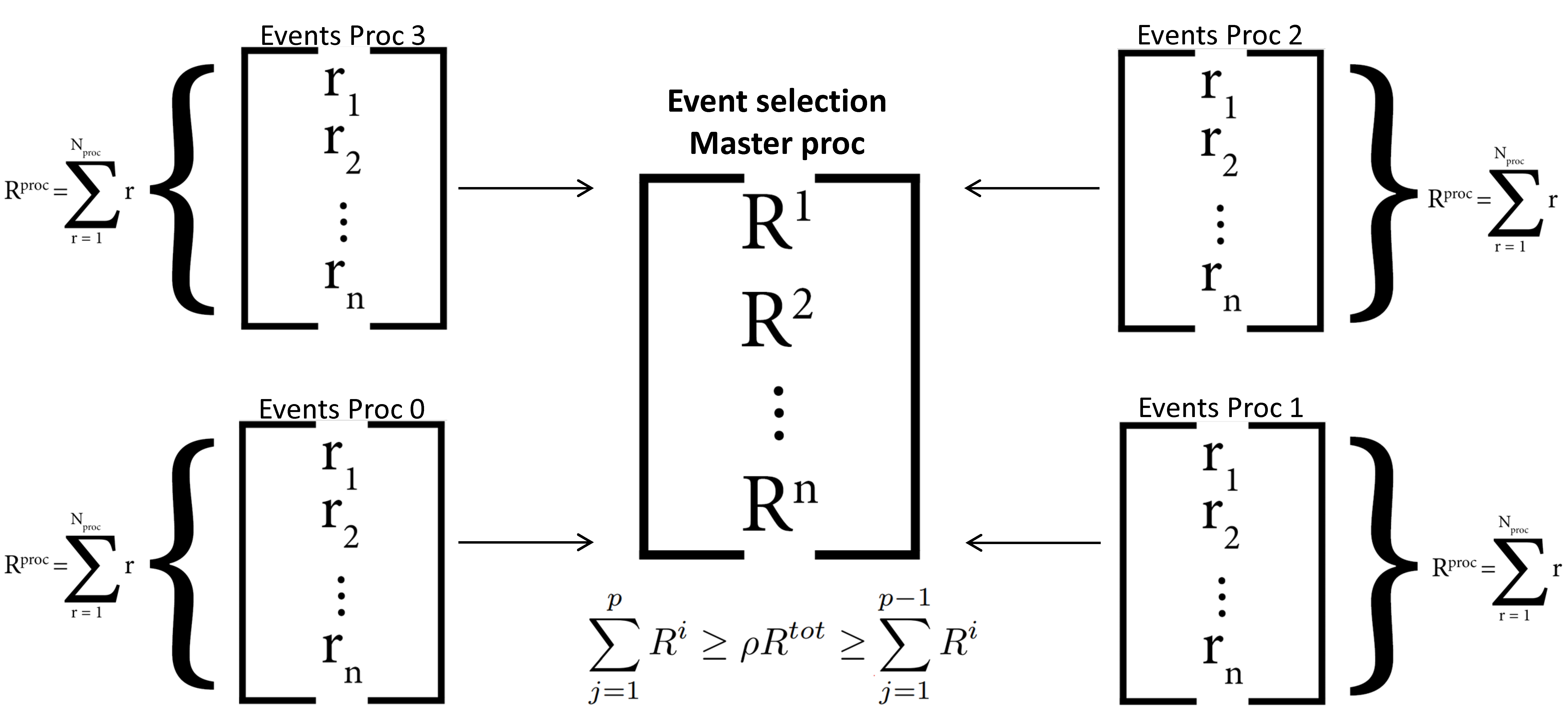
Following, the selected event MPI processor chooses a (local) event for execution (using a rejection-free selection process [1], [2], [3]):
Finally, event data are communicated from the event MPI processor to all other processors and the event is executed globally.
Providing a fully parallelized scheme for event execution within the kMC stage of PAPRECA will be prioritized in future version of the software.
For additional information regarding the rejection-free kMC scheme, as well as the execution/detection of predefined kMC events please see Ntioudis et al. [1], [2] and Fichthorn and Weinberg [3]. For an overview of the LAMMPS software please see Thompson et al. [4].
Supported Predefined events
PAPRECA currently supports 5 different classes of predefined events.
1-2) Reactions (bond-formation and bond breaking). See create_BondBreak command and create_BondForm command.
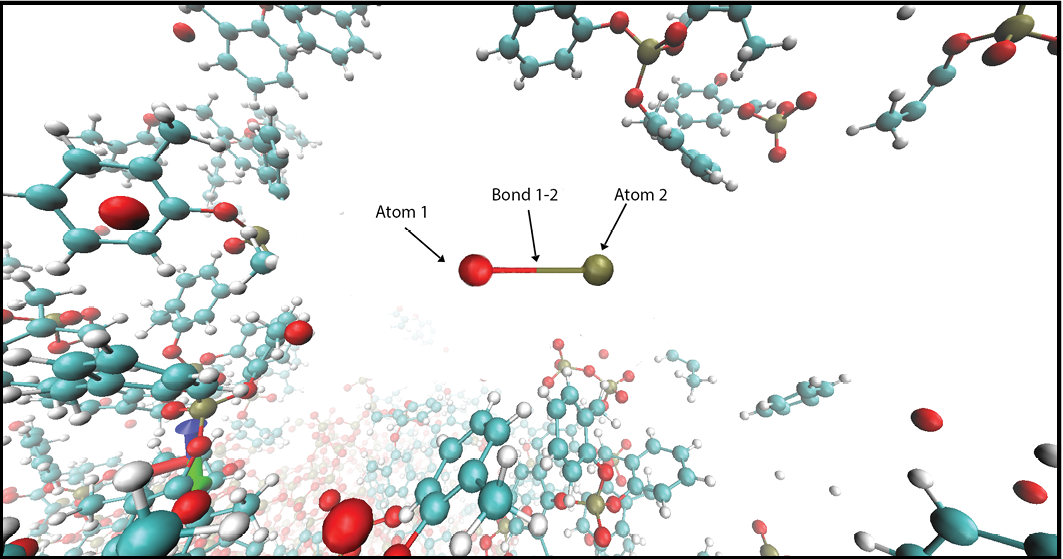
3) Molecular and monoatomic deposition. See create_Deposition command.
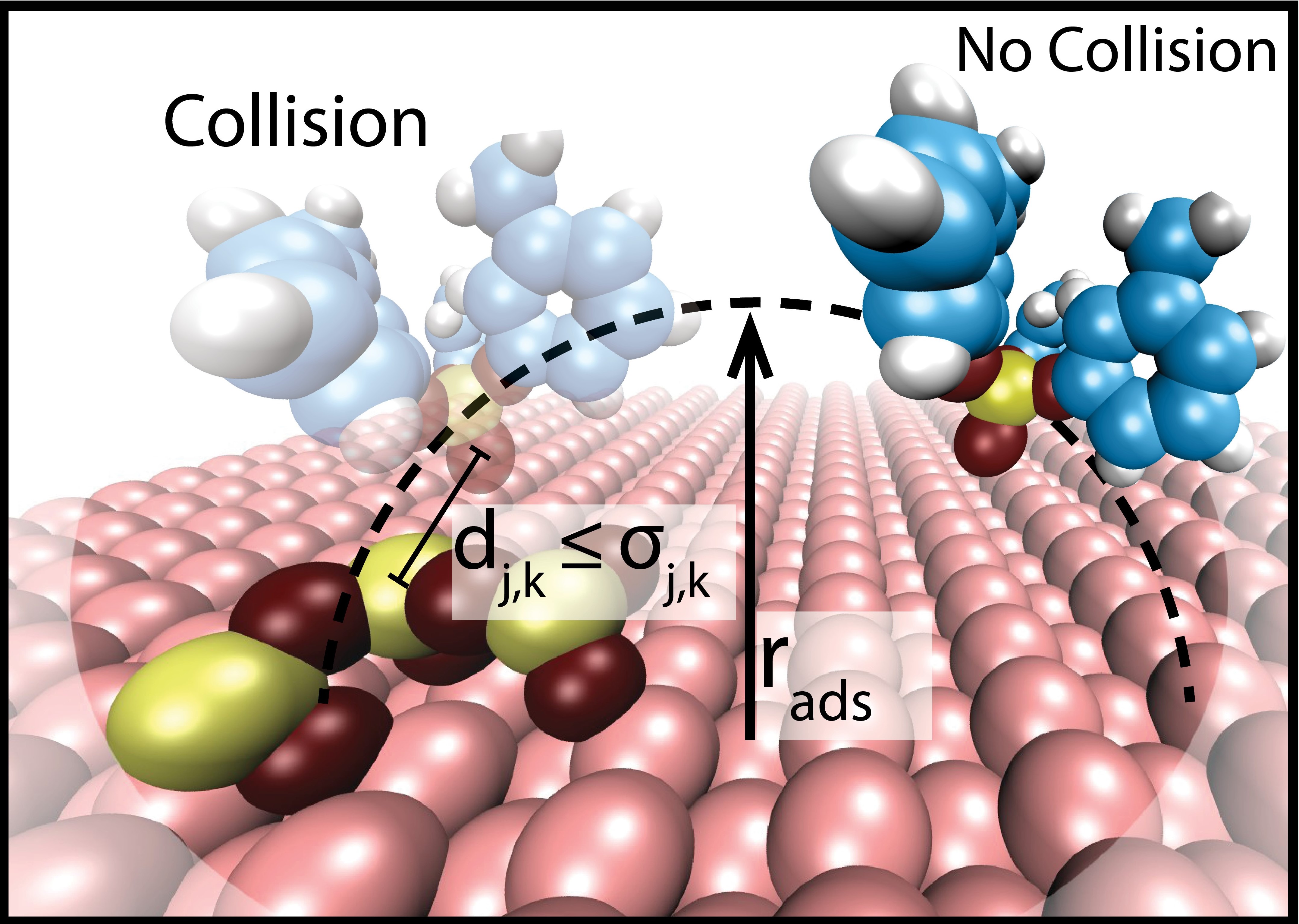
4) Monoatomic desorption. See create_MonoatomicDesorption command.
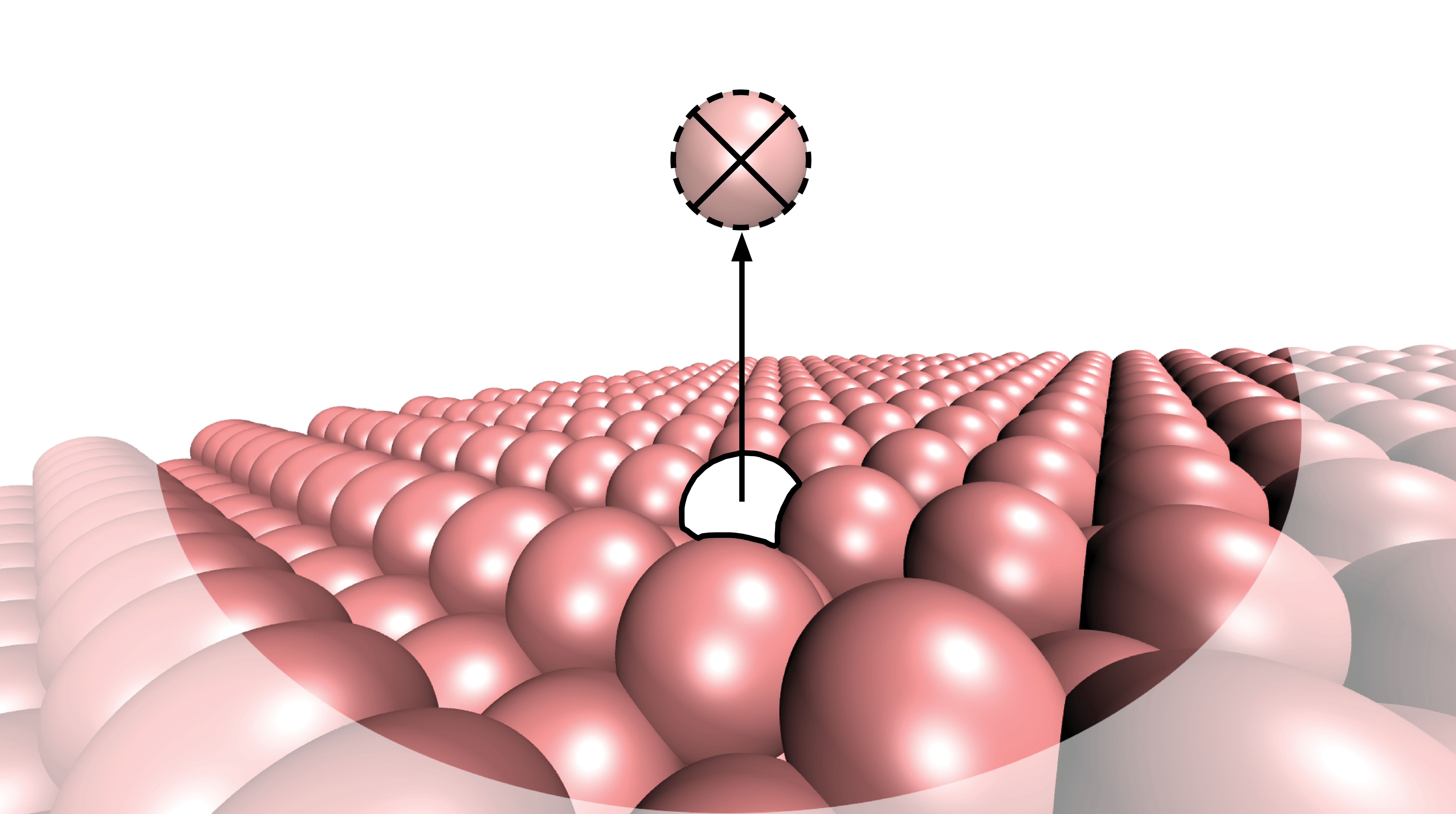
5) Diffusion hops. See create_DiffusionHop command.

Domain Decomposition
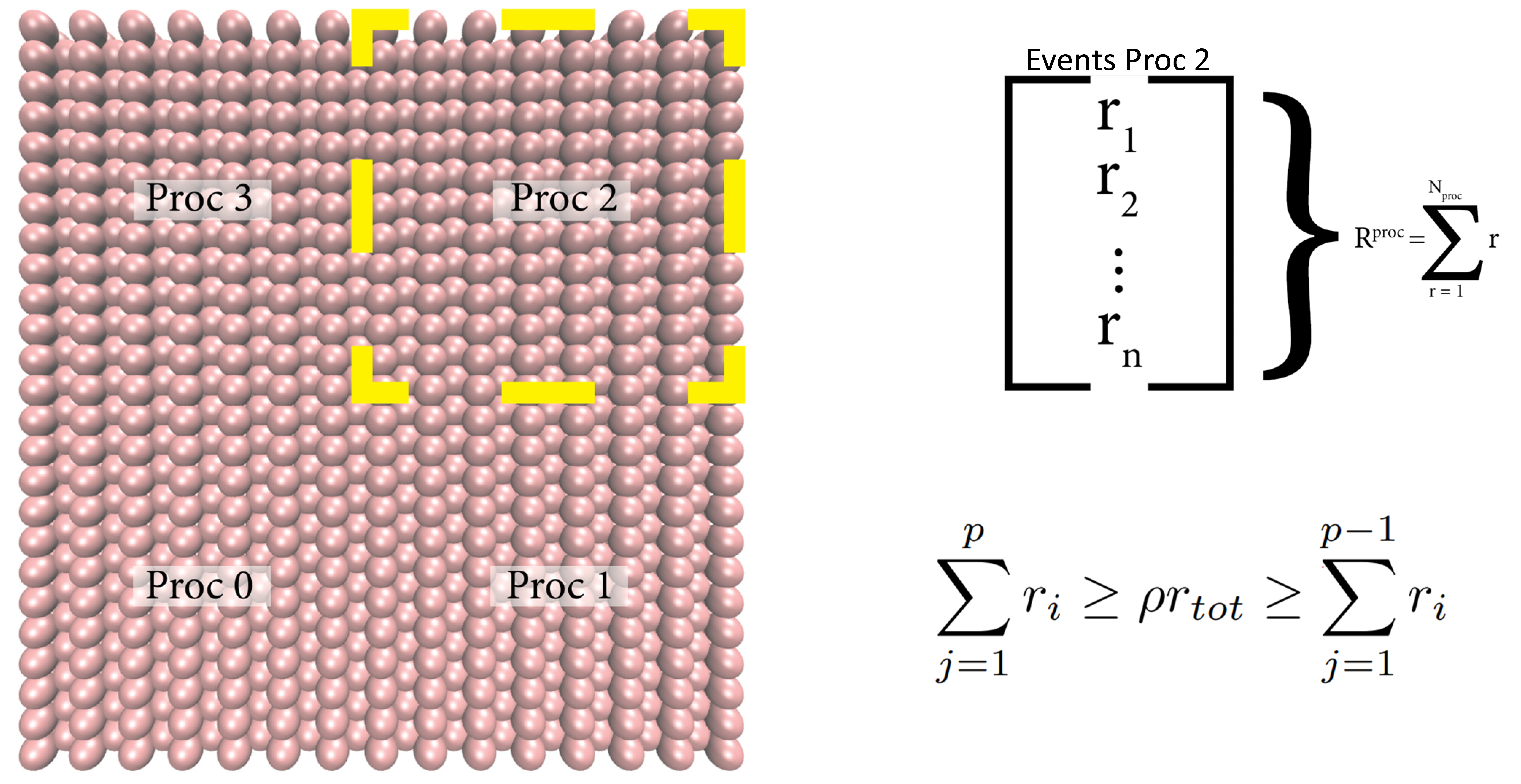
The domain decomposition of PAPRECA is identical to that of LAMMPS. On each kMC stage, PAPRECA searches for local atomic scale processes by accessing whether an owned atom (by a specific MPI process) can be parent to an elementary step, as defined by a predefined template (e.g., a deposition event declared in the PAPRECA input file). For more information regarding the domain decomposition schemes in LAMMPS see this documentation page. To learn more about LAMMPS communication schemes see this documentation page.
Random Numbers and Repeatability
As observed in the general flowchart (see PAPRECA flowchart) PAPRECA fires a single event on each kMC stage. This implies that if the number of MPI processes changes (i.e., you run on 4 instead of 16 MPI processes) PAPRECA runs will not be identical (even if the same random_seed is used in random_seed command). Nonetheless, even in such cases, PAPRECA is expected to produce statistically equivalent trajectories.
PAPRECA and LAMMPS input Files
A PAPRECA run must be set up with 2 input files:
- A LAMMPS input file that initializes the simulation units, the simulation box, the periodic boundaries, and creates the initial system configuration (e.g., starting atoms, bonds, etc.). Furthermore, the LAMMPS input file controls any simulation parameter related to the MD stage of the hybrid kMC/MD run (e.g., thermostats, interatomic potential style, interatomic potential coefficients, etc.). A list of the acceptable LAMMPS s can be found here.
- A PAPRECA input file that sets up the kMC stage of the hybrid kMC/MD run. More specifically, the PAPRECA input file initializes templates for the predefined events of the kMC stage. In the current version, the following predefined events are supported: Bond-formations (see create_BondForm command), Bond-breaks (see create_BondBreak command), Depositions (see create_Deposition command), Monoatomic Adsorptions (see create_MonoatomicDesorption command), and Diffusions (see create_DiffusionHop command). For the commands page for the PAPRECA commands documentation.
Examples of LAMMPS/PAPRECA input files can be found in the ./Examples folder of the parent PAPRECA GitHub repository.
Before you start, it is very important that you familiarize yourself with LAMMPS. See this link from the LAMMPS documentation pages.
Unsupported Features and illegal commands in LAMMPS input files.
- The current version of PAPRECA supports only 3D applications (i.e., no 2D applications are supported).
- The following group names are reserved by PAPRECA and cannot be used in the LAMMPS input file: del_atoms, delete_atoms, deletion, new_mol, fluid, frozen.
- PAPRECA uses a dummy group with id=1 to perform bond deletions (see this LAMMPS wrapper function for more information PAPRECA::deleteBond()), which means that you should NOT use this bond id to define some another bonded interaction. Please see the "kmc.lmp" file located in ./Examples/Phosphate Film Growth from TCP on Fe110/ for an example demonstrating how you can define multiple bond types.
Dealing with large log.lammps files
PAPRECA alternates between kMC and MD stages (performed in LAMMPS). At the setup phase of the MD stage, LAMMPS appends various information on the log.lammps file (e.g., LAMMPS version, MPI warnings, LAMMPS inputs). Additionally, LAMMPS outputs minimization and trajectory information during the MD stage. This means that log.lammps files can become very large. A possible way to stop LAMMPS from constantly writing on the log.lammps file is by adding the following line to your LAMMPS input file (see the relevant LAMMPS documentation page:
Note that the above command will not turn off screen outputs.
Running a PAPRECA simulation
After creating your LAMMPS and PAPRECA input files, a PAPRECA run can be performed by running the papreca executable (as built in your build folder) with MPI.
Examples:
Important Note1: Always use the -in flag to properly pass the input files to the papreca executable.
Important Note2: PAPRECA reads the LAMMPS input file and then the PAPRECA input file. Hence, make sure the LAMMPS input file is provided first in your MPI execution command (e.g., mpiexec or mpirun).
fix papreca
Running PAPRECA requires that the following line is added to your LAMMPS input file (after defining the simulation box):
Of course, using this fix demands that LAMMPS is compiled with the papreca fix (see installation). For more information regarding this fix please refer to fix papreca command.
PAPRECA units
Units within the MD stage are consistent with units as defined in the LAMMPS input file. Units in LAMMPS are set by the units command.
Units within the kMC stage are consistent with units as defined in the LAMMPS input file. Nonetheless, event rates (in frequency units) are internally converted to 1/s. This is done to facilitate the passing of intuitive rate values from the PAPRECA input file to the source code (using the rate_arrhenius, rate_hertz, rate_manual options in the create_BondBreak, create_BondForm, create_Deposition, create_DiffusionHop, and create_MonoatomicDesorption commands). For that reason, PAPRECA will always report time in seconds on the terminal and in any exported file (e.g., heightVtime.log or execTimes.log).
The PAPRECA simulation accounts for both the time elapsed within the MD stage (calculated as the product of the trajectory duration and the timestep) as well as the elapsed time within the kMC stage.
Also, be careful when using the rate_arrhenius, rate_hertz, and rate_manual options (see create_Deposition command) to set a predefined event template rate. You should always use units as requested by the respective rate calculation option.
Important Note: PAPRECA supports the "lj" LAMMPS unit style. However, if you use "lj" units the reported (BY PAPRECA) time will not account for the elapsed time in the MD stage of the run (i.e., solely the kMC stage time will be considered). This happens because there is no straightforward way of converting time units from "lj" to seconds.
Bibliography
[1] Ntioudis, S., et al. "PAPRECA: A parallel hybrid off-lattice kinetic Monte Carlo/molecular dynamics simulator", Journal of Open Source Software, 9(98), 6714 (2024). https://doi.org/10.21105/joss.06714
[2] Ntioudis, S., et al. "A hybrid off-lattice kinetic Monte Carlo/molecular dynamics method for amorphous thin film growth", Computational Materials Science, 229, 112421 (2023). https://doi.org/10.1016/j.commatsci.2023.112421
[3] Fichthorn, K.A. and Weinberg, W.H. "Theoretical foundations of dynamic Monte Carlo simulations." Journal of Chemical Physics, vol. 95, 1991
[4] Thompson, A.P. et al. "LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales." Computer Physics Comunications, vol. 272 2022